387 87 11 11
Víc než nemocnice.

Spinální svalové atrofie (SMA) jsou skupinou dědičných chorob, které se klinicky projevují postupně se zhoršující svalovou slabostí a následnou atrofií, tj. zakrněním postiženého svalstva. Vzhledem k tomu, že se jedná o různorodou skupinu onemocnění, míra postižení bývá různá. Pacienti mají problémy s chůzí, pohybem končetin, polykáním a v neposlední řadě i dýcháním, což je u nejtěžších typů SMA hlavní příčinou úmrtí.
V české populaci se vyskytuje přibližně u 1 ze 6000 narozených dětí, řadí se tedy mezi vzácnější onemocnění. Ovšem i přesto je SMA jednou z nejčastějších příčin úmrtnosti kojenců na vrozené onemocnění.
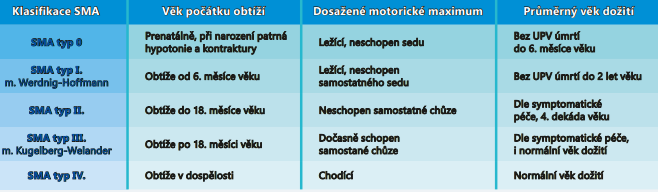
V běžné praxi se v 95 % případů setkáváme s atrofií, která postihuje proximální svaly, tj. svaly nacházející se nejblíže středu těla (svaly hrudníku a břicha, paží, stehen, kyčelní a ramenní pletence). Tyto typy onemocnění jsou způsobené mutacemi genu SMN1, takže je lze diagnostikovat pomocí genetických testů. Klinicky rozdělujeme SMA na 5 typů, a to podle věku, kdy se začaly projevovat obtíže, a dosaženého pohybového maxima (viz tabulka).
Z výše uvedené tabulky vyplývá, že v novorozeneckém věku se můžeme setkat okamžitě po narození se SMA typu 0 nebo častěji se SMA I. typu. V klinickém obraze je hlavním příznakem hypotonie (snížené svalové napětí) se svalovou slabostí, která se rozvíjí většinou záhy po narození. Často se objevují i fascikulace (záškuby) jazyka. Dochází k poruchám polykání, děti nelze krmit a pacienty je třeba vyživovat prostřednictvím sondy. V psychomotorickém vývoji dítěte dochází k jasnému opoždění v motorické složce, sociální kontakt je velmi dobrý a intelekt je zachován. Děti se SMA typu 0 nebo I nikdy nejsou schopny samostatného sedu a bez umělé plicní ventilace většina z nich do 18. měsíce věku umírá na respirační selhání.
Samotná kauzální terapie (=cílená terapie) spočívá v genové terapii, při níž se buď upravuje transkripce genu SMN2, nebo se nahrazuje chybějící gen SMN1. Léčbu SMA provádí čtyři neuromuskulární centra (ve Fakultní nemocnici v Motole, Fakultní nemocnici Brno, Fakultní nemocnici Ostrava a Fakultní Thomayerově nemocnici v Praze) a z toho dvě centra jsou zároveň i Evropská centra pro vzácná onemocnění (Neuromuskulární centrum Fakultní nemocnice v Motole a ve Fakultní nemocnici Brno).

Rychlost zahájení léčby je zásadním prognostickým faktorem, jelikož včasné zahájení léčby, dříve než se rozvinou klinické obtíže, umožní normální vývoj motorických funkcí pacienta.
Po dvouleté pilotní fázi, od 1. ledna 2024, se rozšířil standardní novorozenecký screening o dvě vzácná geneticky podmíněná onemocnění – spinální svalovou atrofii (SMA) a těžký kombinovaný imunodeficit (SCID).
Novorozenecký screening se standardně odebírá mezi 48. až 72. hodinou věku dítěte pomocí odběru kapky krve na 2 propíjecí karty. Na rozdíl od 18 ostatních onemocnění, které dokáže novorozenecký screening odhalit, je diagnostika SMA i SCID založena na molekulárně genetickém testování (qPCR), které se dá vyšetřit ze standardně odebírané suché kapky. V případě pozitivního screeningu bezprostředně následuje cílené genetické testování a pohovor s klinickým genetikem, který by měl pacienty nasměřovat do jednoho ze čtyř neuromuskulárních center v České republice, kde dojde k zahájení léčby. K terapii onemocnění je třeba zaujmout komplexní přístup. Kromě kauzální terapie je nutná intenzivní rehabilitace, nutriční terapie, ortopedická terapie a využívání kompenzačních pomůcek.
MUDr. Kristýna Dvořáková, Neonatologické oddělení
O patogenezi, typech spinální muskulární atrofie (SMA) a další teorii již bylo mnohé řečeno v článcích mých kolegyň z genetiky a neonatologie. Nyní se na toto onemocnění pojďme podívat čistě z hlediska praxe dětského neurologa.
Ještě před necelými deseti lety byla SMA pro každého dětského neurologa velmi smutnou kapitolou; příčinou byla hlavně neexistence léčby zaměřené na příčinu nemoci. Nejčastějším typem je dle původního dělení SMA 1. typu – většinou jde o dítě ze zcela nekomplikovaného těhotenství, porozené v termínu, s dobrou poporodní adaptací. Kolem tří měsíců věku si však rodiče či praktický lékař začnou všímat toho, že je dítě nápadně klidné, jeho chudé spontánní hybnosti a nedostatečných příbytků na váze. Tento stav se prohlubuje a dítě se, řekněme, kolem pátého měsíce života dostane do rukou dětského neurologa. Je chabé, se slabým pláčem, leží na zádech s minimální hybností končetin a jediné, co na něm „hraje“, jsou jeho chytré, zvídavé, veselé oči. Všechny děti se SMA mají totiž zcela normální intelekt. Do roku 2016 byl pro dětského neurologa pohled na takové dítě žalostný. Dostupná byla jen podpůrná a symptomatická léčba. Tyto děti bylo možné rehabilitovat s cílem podpořit jejich hybnost co nejvíce, mohli jsme řešit infekční komplikace, dechové obtíže, aspirace, skoliózu i jiná onemocnění pohybového aparátu. Tlumili jsme bolesti a v pozdních fázích dle přání rodičů či samotných pacientů mohly děti využívat domácí umělou plicní ventilaci.
Od roku 2016 se nejdříve v zahraničí a velmi záhy i u nás začaly objevovat SMA modifikující léky – v roce 2017 přišel Nusinersen, v roce 2020 Onasemnogen abeparvovek a v roce 2021 Risdiplam. Každý z těchto léků přitom funguje jinak. Nusinersen a Risdiplam ovlivňují funkci genu SMN2, a tím částečně přebírají úlohu genu SMN1. Onasemnogen abeparvovek je genová léčba, kterou se do těla vpravuje plně funkční kopie SMN1 genu.
Jakmile na scénu přišla kauzální léčba, v odborných kruzích se začalo intenzivně diskutovat o nutnosti časného, nejlépe presymptomatického záchytu dětí se SMA. V léčbě spinální muskulární atrofie totiž platí, že co je jednou ztraceno, nelze získat zpět. Lékaři z neuromuskulárních center začali bojovat za to, aby byla SMA zařazena do novorozeneckého screeningu. Šlo v podstatě o jedinou šanci, jak zachytit děti bez významných klinických příznaků. Protože se však jednalo o první chorobu, jejíž screening souvisel s genetickým testováním novorozence, nebyla cesta ke schválení a zařazení SMA do novorozeneckého screeningu vůbec jednoduchá.
Od 1. ledna 2022 do 31. prosince 2023 probíhal v rámci České republiky pilotní projekt screeningu SMA u novorozenců. Screening byl dobrovolný a matky novorozenců musely podepsat souhlas s genetickým vyšetřením dítěte. V rámci tohoto projektu bylo celorepublikově zachyceno dvacet dětí. Dva další pacienti byli, kvůli jejich těžkému klinickému stavu ihned po narození, zachyceni již před novorozeneckým screeningem, jeden minimálně příznakový pacient byl zachycen jako starší sourozenec dítěte pozitivního ze screeningu. Já osobně považuji záchyt dvaceti dětí za obrovský úspěch moderní medicíny. Deset dětí ročně dostane šanci na diametrálně lepší a kvalitnější život, než kdybychom čekali na rozvoj klinických příznaků. Od 1. ledna 2024 byla SMA oficiálně zařazena do novorozeneckého screeningu.
Tato zásadní změna nebude znamenat, že zcela vymizí děti, u nichž se příznaky SMA již klinicky vyjádřily. Budou případy, kdy děti screeningu uniknou (např. při domácích porodech, z vůle rodičů, děti imigrantů apod.). Děti, které byly genovou terapií léčeny na počátku její dostupnosti u nás, jsou dnes již tří- až čtyřleté a jsou schopné chodit a vyvíjet se tak, že by ani poučený laik nepoznal, o jak závažně nemocné děti se jedná. Pacienti s identickou diagnózou by před érou genové léčby nikdy nedosáhli samostatného sedu a v předškolním věku by umírali.
Důležité je ještě zmínit, že genová léčba není určena pro všechny děti. Existuje velká skupina pacientů, kteří se narodili před jejím zavedením na trh a jejich postižení jsou v tuto chvíli již tak těžká, že tato léčba indikována není. Druhou malou skupinkou jsou novorozenci se SMA, kteří trpí těžkým postižením již při porodu. Prognóza těchto dětí je velmi nepříznivá a mnohdy se volí léčba symptomatická či paliativní.
Genová léčba je a do budoucna jistě bude velmi kontroverzním tématem. Je zde ovšem velký předpoklad, že děti, které by dříve v brzkém věku zemřely, budou v dospělém věku aktivními vozíčkáři s plným přínosem pro společnost – a to za mě dává obrovský smysl.
MUDr. Lenka Kabourková, Ambulance dětské neurologie Dětské oddělení
Spinální muskulární atrofie (SMA) je závažné, geneticky podmíněné nervosvalové onemocnění, které se projevuje progresivní svalovou slabostí.
SMA je způsobena chyběním produktu genu SMN1, který je nezbytný pro přežívání motoneuronů (nervových buněk, které řídí příčně pruhované svaly těla a tím náš volní pohyb). Pokud nemají motoneurony v míše dostatek tohoto produktu, odumírají, tím nemohou stimulovat svalová vlákna, vlákna se ztenčují až mizí. Tak název nemoci přesně vystihuje problém – míšní (spinální) nervové buňky hynou a svaly, které tyto nervové buňky řídí, se následně ztrácejí (svalová atrofie). Člověk ztrácí možnost volního pohybu. Vše se děje postupně, v různém tempu, ale bez léčby se nemoc nedá zastavit.
Gen SMN1 – recept, podle kterého naše nervové buňky „vaří“ svou výživu – máme všichni ve dvou kopiích. K rozvoji nemoci dochází pouze při vyřazení obou kopií SMN1 genu. Tzn. že i jedna funkční kopie vytvoří pro míšní nervovou buňku dostatek produktu a na člověku není jedna poškozená kopie nijak poznat – takový člověk s jednou poškozenou kopií je zdravým přenašečem SMA. Přenašečem je v české populaci přibližně každý čtyřicátý z nás. Pokud se potkají dva takoví zdraví přenašeči, mohou, ale nemusí mít potomka s SMA – oba rodiče musí pro rozvoj nemoci předat svému dítěti tu svou poškozenou kopii genu SMN1, dítě má pak obě kopie poškozené a někdy v životě začne mít problém. Čas nástupu onemocnění ovlivňuje jiný gen (SMN2), který u člověka s funkční alespoň jednou kopií genu SMN1 není vůbec důležitý, ale při vyřazení obou kopií spoluurčuje formu nemoci (kdy se objeví první příznaky a jak nemoc bude postupovat).
Protože je v současnosti dostupná léčba, která zásadně mění prognózu a kvalitu života pacientů, byla kvůli včasnosti zahájení léčby SMA zařazena do novorozeneckého screeningu. Léčba totiž nedokáže vrátit změny, které se již staly, nevrátí ani nenahradí odumřelé nervové buňky, ale umí zastavit jejich úbytek. Proto je zásadní ji podat co nejdříve, ideálně před nástupem příznaků.
Kromě podpůrné léčby zmírňující příznaky a rychlost postupu onemocnění, jsou v současnosti dva zásadní přístupy, které umí bránit ubytku motoneuronů. Lék, který se opakovaně aplikuje do páteřního kanálu a který využívá pacientův vlastní gen SMN2, aby tvořil „výživu“ pro spinální motoneurony místo poškozeného genu SMN1. Tento lék je schválen prakticky pro všechny pacienty s prokázanou SMA. A pak nejnovější genová terapie, která jednorázovou aplikací vnese do lidských buněk funkční kopii genu SMN1 a tělu nahradí jeho poškozené kopie. Podání genové terapie je však možné jen u nejmenších dětí, a proto má screening SMA a její včasná diagnostika zásadní význam.
Screening probíhá z kapky krve odebrané v porodnici a detekuje pouze případy, kdy chybí obě kopie genu SMN1, tj. asi 95 % případů SMA. Nedetekuje přenašečství. Nedetekuje vzácné situace, kdy jedna kopie genu chybí a druhá je přítomná, ale poškozená nějakým drobnějším „překlepem“. Screening má tedy za cíl odhalit toto závažné onemocnění u většiny pacientů včas jednoduchým, snadno proveditelným testem, který nám nepřináší žádné nevyžádané informace navíc, ale není 100%. Proto má-li lékař na základě příznaků podezření na SMA, musí vědět, že tato nemoc nebyla screeningem úplně vyloučena, a po domluvě s genetikem se může gen SMN1 znovu a podrobněji vyšetřit. V případě, kdy je gen SMN1 v pořádku, přichází v úvahu i jiné, vzácnější vrozené příčiny svalové atrofie.
V případě, kdy oba rodiče vědí, že jsou přenašeči SMA, mohou se rozhodnout jít cestou plánovaného rodičovství a za využití metod IVF (asistované reprodukce) jim může současná medicína pomoci vybrat potomky bez genetické dispozice k SMA. Nebo může žena podstoupit vyšetření plodu v těhotenství a případně těhotenství ukončit. Ani současná léčba nedává 100% záruku, že dítě s SMA nebude mít vůbec žádný problém.
MUDr. Zuzana Šimková, vedoucí lékařka Ambulance lékařské genetiky
Dědičná onemocnění byla dlouho vnímána jako daná a neměnná, ve většině případů neléčitelná. Měnit genetickou informaci ve prospěch pacientů, tedy provádět genovou terapii, se lidstvo pokouší od roku 1990, nicméně často neúspěšně. V posledních několika letech se však karta obrátila a jsme svědky mnoha úspěšných projektů. V současnosti navíc probíhají stovky studií, od léčby fatálních chorob až k pomoci se závislostí na nikotinu.
O nemoci SMA mluvíme tehdy, když vlivem chyb v DNA, a to v obou kopiích genu SMN1, nevzniká funkční „SMN protein“. Situace je ale trochu komplikovanější: kromě dvou kopií genu SMN1 máme ještě dvě kopie genu SMN2 – nefunkční evoluční kopii genu SMN1, odborně nazývanou pseudogen. Geny SMN1 a SMN2 jsou téměř shodné, z celkových 882 se liší pouze ve dvou písmenkách kódující sekvence DNA, z nichž zásadní je změna písmena C na pozici 840, které je v genu SMN2 změněno na T.
Tato změna způsobí, že podle genu SMN2 vzniká jen 10 % funkčního SMN proteinu. A aby situace byla ještě složitější: dvě kopie genu SMN2 má 60 % populace, ostatní lidé mají 1–8 kopií tohoto genu. Čím větší je počet kopií SMN2, tím mírnější jsou příznaky SMA.
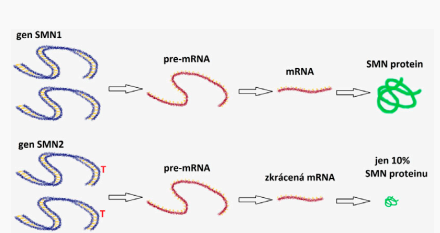
Situace u zdravého člověka bez mutací genu SMN1
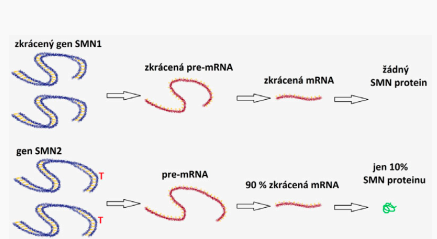
Situace u pacienta se SMA
V jádře buňky se nachází DNA, velice dlouhá molekula skládající se ze dvou vláken, které se skládají ze čtyř „písmenek“ – A, C, T, G. V případě potřeby se část molekuly, gen, přepíše do velmi podobné jednovláknové molekuly, tedy premRNA. Ta se pak „přeskládá“ – hlavně se z ní vystřihnou nepotřebné části – a vznikne mRNA, kratší jednovláknová molekula, podle které se už mimo jádro vyrobí protein neboli bílkovina, jenž má v buňce vždy nějakou důležitou funkci. Většinu genů (molekul DNA) máme ve dvou kopiích, po jedné od každého z rodičů.
Léčit SMA je dnes možné dvěma způsoby, jednak ovlivněním tvorby mRNA u SMN2 pomocí uměle vytvořené nukleové kyseliny a jednak pravou genovou terapií, tedy dodáním nového a neporušeného genu SMN1.
Léčba prostřednictvím ovlivnění tvorby mRNA spočívá v dodání krátkého kousku uměle vytvořené nukleové kyseliny, sestávající z 18 speciálních písmenek, která se v přírodě nevyskytují. Tento kousek se váže na pre-mRNA pseudogenu SMN2 a přinutí ho vyrábět správnou mRNA, ze které pak vzniká funkční SMN protein. Účinek léčby není trvalý a lék je potřeba opakovaně podávat po celý život. V současnosti jsou k dispozici dvě látky, které toto dokáží: nusinersen, první lék na SMA, který existuje od roku 2016 a bohužel se musí podávat přímo do mozkomíšního moku, a novější risdiplam, který existuje od roku 2020 a je již ve formě tablet.
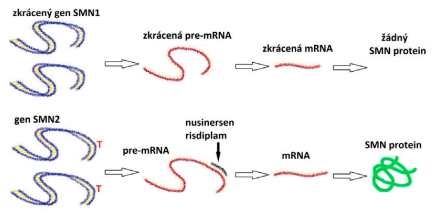
Léčba ovlivněním tvorby mRNA
Genová terapie pomocí funkčního genu SMN1 spočívá v „infekci“ speciálně upraveným virem, který nese sekvenci funkčního genu SMN1. Tento laboratorně modifikovaný virus sestává z DNA několika organismů a je „zabalen“ v obalu viru AAV9. Tento lék, který byl schválen roku 2019, nese téměř tajemný název onasemnogen abeparvovec. Nejedná se však o nic magického – název vychází ze systematických pravidel pro pojmenovávání moderních léků: „onasemnogen“ nám říká, že se jedná o lidský gen SMN a „abeparvovec“ znamená, že je zde silná regulační oblast z β-aktinu, že byl použit virus AAV9 a že se jedná o nedělící se vektor. Tento lék se aplikuje infuzí do žíly jen jednou za život. Virová DNA se dostane do jádra lidských buněk a dále se nedělí ani nezačleňuje do jaderné DNA. Protože se ale cílové nervové buňky rovněž nedělí, zůstává produkce klíčového SMN proteinu ve tkáni stabilní. Od podání léku již nemoc nepostupuje.
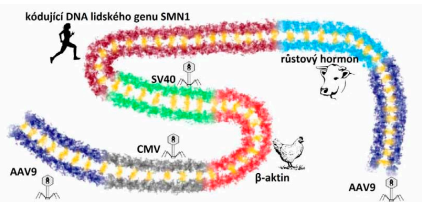
Struktura léku onasemnogen abeparvovek. Lék obsahuje sekvence DNA z následujících: Virus AAV9, díky kterému je možný úspěšný přenos DNA do virového obalu; viry CMV a SV40, kuřecí β-aktin, růstový hormon skotu – regulační oblast zajišťující správný přepis do mRNA a intenzivní produkci SMN proteinu; a hlavně kódující sekvenci lidského genu SMN1.
Mgr. Ondřej Scheinost, vedoucí Laboratoře molekulární biologie a genetiky